

Nature’s neglected GoF laboratory
Nature’s neglected GoF laboratory
The case for a zoonotic origin of SARS-CoV-2


This is a very long article about the origins of SARS-CoV-2. To get a comprehensive picture of why scientists believe a zoonotic origin best explains the evidence, it is worth reading it in full. If you are just interested in specific scientific arguments around genetic engineering or case epidemiology, you can start by looking at Chapter 2, Sections B & C. If you are curious about the big picture and what to do next, you can go to Chapter 3.
Chapter 1: Scientific uncertainty
Background
Ever since the emergence of SARS-CoV-2, scientists have been working to reconstruct how, where, and when this virus jumped into humans and subsequently caused a pandemic. Almost immediately, speculations were raised if SARS-CoV-2 was indeed a ‘natural’ virus or whether it was engineered or cultured in a lab given the location of the outbreak and the unprecedented harm it caused worldwide (Read this for a short history on the conspirational thinking that followed every emerging pandemic).
Today, many competing origin narratives built on top of scientific uncertainty have swept over the globe, fueling speculations, emotional discourse, and even geopolitical tensions.
So let me say a few things about the sideshows surrounding lab leak and hopefully bring the science back into focus:
First, the dynamics of social media amplification and the crowdsourcing of misinformation have wrought havoc on what is supposed to be a precise scientific discussion. Overall, what exactly somebody means when they claim SARS-CoV-2 might have ‘leaked’ from the lab has been a moving target. From ‘bioweapon to hurt Trump’, ‘secret GoF research & pathogen escape’ to ‘unwise serial passage experiments’ or ‘reckless viral modification’ to ‘sloppy bat sampling accident’; the whole spectrum of possible human actions, from intentional release, conspiracy & cover-up or remote accident, has been subsumed under one ‘lab leak’ term. This is of course impractical.
Everybody loses by operating with bad definitions
Second, dealing with this panacea of origin stories has been a frustrating experience for scientists. Rightfully calling some wild scenarios “conspiracy fantasies” gets intentionally misrepresented by motivated actors as ‘disrespectful mocking’ or ‘trying to disparage’ genuine scientific inquiry. On the other hand, granting validity to some potential ‘lab leak’ scenarios and supporting further scientific inquiry is interpreted as the legitimization of the whole cottage industry of opportunistic LARPers, attention grifters, and online trolls. This is unworkable as the noise to signal ratio skyrockets and favors the loudest shouters, not the most prudent analysis.
Third, discussions about future lab safety protocols, potential dangerous research practices, and biosafety are important. However, these discussions ought to be decoupled from the origin question. Even if it is proven beyond doubt that SC2 has a natural origin, everybody should continue to be interested in labs not accidentally causing the next pandemic. Scientists take biosafety and public concerns seriously, they do not need this pandemic to have come from a lab to continue to do so. Let’s not sacrifice truth on the altar of convenience for a different purpose.
Fourth, there have been countless unsubstantiated insinuations of a cover-up by Western scientists or institutions, baseless ad hominems of lab leak proponents towards “Zoonati” scientists, and wild claims using China’s lack of transparency as ‘Carte Blanche’-type of proof for whatever pet theory one fancies. It is worth noting that all of these ‘human investigative’ avenues of inquiry into a ‘cover-up by virologists’ through decontextualized FOIA/emails, as well as reasoning about human motivations, are rightfully disputed, circumstantial at best and ultimately fall short of explaining by themselves how the SARS-CoV-2 viral genome we observe came to be. It’s harassing innuendo, mostly bad faith, and ignores that in the end, a scientific question needs a scientific answer.
So with all these (ultimately irrelevant, as this article will show) sideshows going on, how do we bring discourse back to science?
I’m not sure, but I believe that a lot of focus has been on debunking the conspiracies, showing flawed reasoning or unscientific argumentation structures, with little results. So I’ll try to do something different here:
I’ll explain, as simply as I can, why virologists believe a zoonotic origin scenario (and there are a few variations as well) is far more likely and better supported by scientific evidence than any and all ‘lab leak’ origin scenarios.
For that, we will have to focus on the evidence and the scientific uncertainty surrounding the following core areas:
risk of zoonotic spillovers/lab pathogen escapes
the genomic features of SARS-CoV-2
the early epidemiology in Wuhan
the coronavirus lab work performed by scientists
It is this large body of emerging and established scientific evidence relating to these points that indicates a very strong likelihood (not yet definitive proof) for a natural origin of SARS-CoV-2 for most domain experts, the wider scientific community, science journalists, and other scientific institutions.
But before we get to the evidence, we need to understand how exactly scientists deal with uncertainty in the first place.
On Bayesian Reasoning
In the absence of clear scientific proof, or direct and compelling evidence, scientists use a Bayesian probability framework to assess the likelihood of competing hypotheses. The guiding question is: How likely does my hypothesis explain (all!) the relevant data compared to competing hypotheses?
Let’s take an example:
We observe that the street outside is wet. We have three competing hypotheses: First, it rained. Second, the street cleaners washed it. Third, the neighbor splashed it accidentally while watering his nearby garden, as has happened before. Baseline likelihoods would go somehow like this: It rains on average once a week, so odds on any particular day of the street being wet is 1/7. Street cleaners come once a week too, 1/7. The neighbor waters his garden also once a week 1/7. So each scenario has about the same likelihood of being true. Now upon consultation of our calendar, we found out that it’s Sunday, reducing the likelihood of street cleaners dramatically. We also discovered that yesterday’s newspaper predicted rain for the night, and remembered that our neighbors were thinking about going on holiday this month, albeit we forget when exactly. Our updated likelihoods might then look somewhat like this: Rain ~95%, Neighbor ~5%, Street cleaners <1%. Without further evidence, this is the best we can do. We don’t have absolute certainty, yet most reasonable people would agree that blaming the street cleaners or even the neighbor might be premature at best, and highly irresponsible given the data.
This is how Bayesian reasoning allows us to estimate likelihoods when comparing different hypotheses. Science tries to reduce uncertainty as much as possible, but not more than is warranted. Note also that there are several caveats to Bayesian approaches. For example, unknown variables or new hypotheses might completely change the game. While the odds of kids playing with water pistols outside might be negligible for any given day, if we were to observe a forgotten water pistol on the wet street, we’d have to update our likelihood estimates too. So don’t be fooled by some non-expert influencer claiming to have done Baysian analysis, odds are they are missing many important variables (& considerations) because they just don’t know about them given their lack of expertise in the matter.
In the case of SARS-CoV-2 emergence, multiple variables with prior unknown likelihoods (see next chapter) have to be navigated to narrow down the widespread uncertainty surrounding potential origin hypotheses.
However, the more we learn about each variable’s true likelihood through the accumulation of evidence, the better we can assess and compare the strength of each competing origin hypothesis. Another way to phrase it is that when one has two equally parsimonious hypotheses, whichever best predicts all evidence is the most likely to be true. This is why we have to look at all the evidence, together.
Science tries to reduce uncertainty as much as possible, but not more than is warranted.
Alright, you get the point. Bayesian hypothesis testing is very common for scientists, especially when a null hypothesis cannot or ought not to be defined. More philosophically, one could describe science as a Baysian effort that tries to approximate what is most likely ‘true’ (as in ‘real’) given the current evidence. All while maintaining an open door for more data to come in.
To finish this section, one more point:
Scientific likelihoods estimates (Bayesian or not) are not a gamble, guessing game, or opinion, nor can they be discarded as such. They are the objectively best current estimates for the true shape of reality. They matter, a lot, for decision-making and collective action. The overarching goal of this article is to develop an understanding that while not everything can be determined with absolute certainty by science, having alternative hypotheses around which are not (yet) disproven does not imply they are on equal footing or should influence our decision making. We’ve already mentioned that the current evidence is not sufficient to completely prove or rule out all possible natural origin or lab leak scenarios, nor is it likely that such incontrovertible evidence will soon emerge. And yet, we have to act and make decisions today.
But let’s start with the evidence and see where the scientific uncertainties really lie, shall we?
Chapter 2: The Science
Section A) What are the likelihoods of zoonotic spillover from an animal reservoir vs. a lab leak infecting humans?
Animal reservoirs
Bats serve as the biggest animal reservoir for CoVs, and exactly how big and diverse this reservoir might be has been subject to continued study. Of the ~5000 mammalian species on earth, over 1000 are bats. However, while bats might carry CoVs, they are by far not the only species that can get infected by them, and they are also not the most likely to come in contact and directly transfer it to humans.

The last two CoVs predating SC2 which caused an epidemic in humans were infections through intermediate animals (civet cats and camels for SARS1 and MERS, respectively). CoVs in bats do not only infect the respiratory system but are often more prevalent in the gastrointestinal (GI) tract or kidneys, allowing for bat Guano and urine to serve as primary routes of infection in cave feeding animals like Civets, or infect human workers collecting Guano fertilizer. Overall, bats CoVs have been considered the original reservoir of many human disease-causing pathogens and will continue to pose a risk to our health (further reading: here).
Viral diversity
It is hard to come by estimations of just how many viruses with zoonotic potential are out there, but it is certainly in the order of tens of thousands. Recent surveillance of nearly 2000 game animals in China revealed 102 animal-infecting viruses, among them 65 previously undiscovered and 21 considered high risk for human infection.
Specifically for CoVs, multinational efforts to surveil and assess risks of CoVs in many susceptible nations established a wide viral variety of CoVs in bats. The vastness of viral diversity in the wild is important to appreciate because we might be blind to many potentially pathogenic viruses that pose a real and immediate danger to us as they exist today.
Before SARS-CoV-2, researchers were focusing on batCoVs that were genetically quite related to SARS, believing that these cousins were most likely to emerge in humans. Since then, we’ve learned that there are indeed genetically very different viral lineages capable of infecting humans, and the whole weight of that threat becomes palpable considering that these different lineages actively mix and share genomic puzzle pieces through recombination, which is what happened with the mosaic genome of SC2.
We will look deeper into what these puzzle pieces are when assessing the genomic features of SARS-CoV-2 in the next section [B].
Zoonotic opportunities per year
It is not a question of whether more zoonotic jumps of dangerous viruses into humans will happen in the future, only when they will hit us.
The first point to mention here is the vast geographic range of bat species hosting SARS-related (SARSr) viruses. Bats are living three times longer than mammals of similar size, are known to migrate several hundreds of kilometers (world record: 2224 km), and share cave habitats with many other bat species. Kind of the animal equivalent of a cosmopolitan.
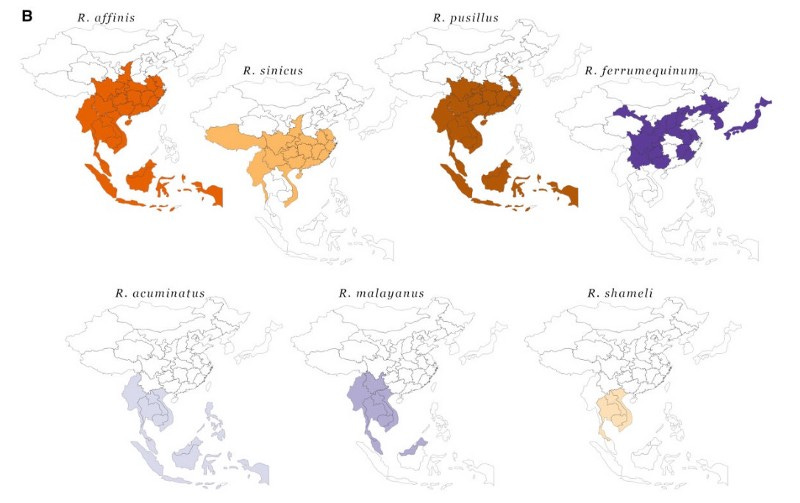
More importantly, serological surveils found that up to 4% of people living close to bats or working closely with wildlife in southern China were infected with dangerous animal-borne viruses, including CoVs. Antibodies against CoVs had also been found in one out of five people who’d had direct contact with bats and other wildlife in Laotian provinces where the currently closest (in time) ancestor of SARS-CoV-2 was found. A recent modeling study suggests that overall, tens of thousands of zoonotic SARSr-CoV infections happen every year in Southeast Asia. Luckily for us, most human infections with a virus even as ‘well-adapted’ to humans as SC2 would die out 95–99% of the time in these low-density rural areas where they occur. In denser cities, it is a different story.
In the end, zoonosis is about opportunity. The more chances we give viruses to jump into humans, the more lottery tickets we allow them to buy, the more likely a well-adapted and dangerous virus will eventually find itself ‘winning’ the genetic jackpot; causing another pandemic.
Given the current trend of ever-increasing mobility and transportation, mostly unregulated wildlife trade, population growth, and deforestation (among other encroachments into natural habitats) we can expect more animal pathogens to cross over to humans in the future if nothing changes.
In the leadup to the SC2 pandemic, a different pandemic of African swine flu virus (ASFV) decimated pork supply in China which lead to alternative sourcing of meat, including from wildlife and fur farms. Economic demand has always been a powerful force to disrupt ecosystems, and it is easy to picture how changes in demand increased opportunities for zoonosis spillover in China even beyond the already high constant risk at a time right before the pandemic started.
Laboratory escapes
The vast majority of labs around the world do not work with dangerous pathogens. There are about 60 labs worldwide who handle the most dangerous pathogens, and probably around ~500 biocontainment labs (no absolute number available) work with potentially dangerous biological materials (bacteria, viruses, fungi, transformed cells, infected tissue cultures, contaminated samples of blood/urine). Only a minor fraction of these labs work on pathogens that cause risks to wider society if containment is ever breached.
Lab accidents and the unintentional escape of pathogens from these facilities have happened before. However, these events are rare enough that they get their own Wikipedia page and plenty of news coverage (consider the list includes not only viruses but other pathogens as well, it also includes suspected but not confirmed events like the 1977 Russian flu). Most pathogen escapes are accidents (self-stabbing with a syringe, animal bites, or equipment failure) causing singular infections that are often self-reported and self-contained. This makes intuitive sense, given that the scientists who got bitten or stabbed themselves might pay extra attention to symptom onset. Some accidents are the result of misconduct and individual error causing containment breaches. Most pathogen research investigates existing pathogens and manipulates specific features of those pathogens to address targeted scientific questions. There have been no laboratory-associated outbreaks of previously unknown or novel pathogens. In the case of SC2, it is also dubious whether an uncharacterized viral pathogen would ever serve as the basis for engineering or could be created de novo, even if scientists wanted to. (More on that in section B as well)
Conclusion Section A:
Research activities involving dangerous pathogens can lead to accidental infections or ‘leaks’ of that pathogen. While it is difficult to come up with hard numbers for how often “pathogen escapes” happen, these incidences are certainly a few orders of magnitude less common than zoonotic jumps from nature. Furthermore, lab escapes first require that the labs are in possession of that pathogen, and engineering work performed on uncharacterized pathogens is so far unheard of. No previously unknown or novel pathogens have ever been associated with a lab escape.
On the other hand, the vast majority of all human viruses, CoVs or not, trace back to some animal reservoir. All previous human-infecting CoVs have had a zoonotic origin, from SARS to MERS and including the four endemic HCoV strains. Bats stand out as a mammal family because of historical precedent and from what we learned through genomic surveillance. This is not new knowledge, in fact, scientists have been sounding the alarm about bat reservoirs for years before SC2 started a pandemic.
We know, without any doubt, that nature’s dangerous GoF laboratory comes up regularly with very unique viruses capable of infecting other animals, including humans. Putting the estimates from bat numbers, geographic range, serological surveys, and human encounters together shines a devastating light on the vast opportunities for zoonosis we offer these viruses. It is not a question of whether more zoonotic jumps of dangerous viruses into humans will happen in the future, only when the next pandemic-ready natural virus will hit us.
But is SC2 a naturally evolved virus?
Section B) What are the likelihoods that a viral genome just like SARS-CoV-2 could arise in nature vs. engineering in a lab?
Was the viral genome backbone ‘created’?
The whole SC2 genome consists of roughly 30.000 nucleotides and scientists have been investigating with a fine-tooth comb whether any signs of engineering can be discerned. Understanding all the methods can get a bit technical, but it is worth mentioning at least some. Obvious signs of engineering would be sequence or protein tags, or really any stretch of nucleotides that are known to not be of viral origin. None were found. In fact, after the pandemic started, researchers went out sampling more CoVs in wild bats and found SARS-related beta coronaviruses (Sarbecoviruses) that had very high nucleotide similarity (> 97–99% depending on ORF segment), meaning these viruses would only differ by a few sporadic single mutations every few hundred nucleotides.
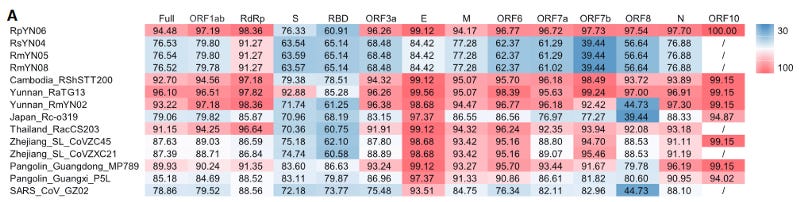
These wild viruses serve as direct evidence that the SC2 backbone was not engineered from RaTG13 or any other known virus. One reason is that nobody would randomly engineer synonymous nucleotide changes hundreds of times all over the genome. Another is the ‘coincidence’ of these engineered nucleotides matching the ‘ancestral’ nucleotides at that position from wild viruses which were not yet sequenced. Earlier recombination analyses had already disproved the idea that SARS-CoV-2 could’ve evolved from RaTG13, purposefully or accidentally either. (further reading: here) Starting from RaTG13 (or any other known CoV) leaves no plausible path to end up with SARS-CoV-2. On top of that, no unusual frequency or distribution of nucleotide transitions/transversion, k-mers, or dN/dS ratio was found either which could point to any conceivable manipulation like cell culture or mutagenic agents.
All these lines of evidence unequivocally indicate that the overall SC2 viral genome is naturally evolved. But could this natural virus have been ‘heated up’ through engineering?
Was binding affinity to hACE2 optimized?
Any proposition that humans ‘needed to optimize’ a genetic feature because it apparently “functioned so well” is ignorant and fundamentally misunderstands the power of selection.
Another point that was often raised was the alleged ‘unnatural adaptation’ human ACE2-receptor binding domain (hACE2-RBD) of SARS-CoV-2. First, this argument from apparent ‘optimal function’ is unfortunately unscientific (akin to creationism), given that natural selection can and has produced remarkable functional feats all around us. Perceived optimality is irrelevant. Second, it is also not true, because nothing about SC2 RBD is ‘optimal’, just good enough at infecting human cells to establish human-to-human transmission chains. Since then, many acquired mutations in the RBD have evolved that increase binding affinity to hACE2, given that we allowed the virus to run rampant in humans.
But higher affinity is not the only factor in how the RBD got more ‘optimized’ for human-to-human transmission through selective pressure. Just how much the RBD changed and evolved since it entered humans is remarkable. (and don’t get me started on the RBD mutations in the Omicron variant)
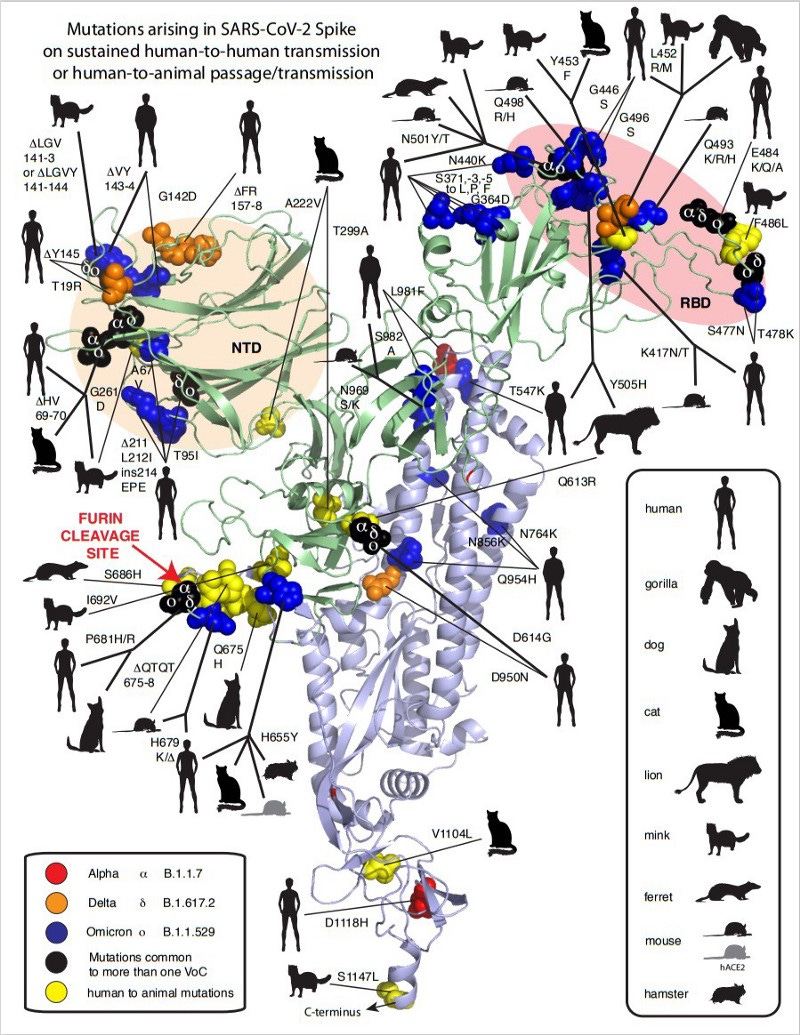
Any proposition that humans ‘needed to optimize’ a genetic feature because it apparently “functioned so well” is ignorant and fundamentally misunderstands the power of selection.
However, probably the best proof that nature can come up with great human-binding RBDs was found mid-2020 in Laos. During a bat sampling expedition, French & Laotian scientists found SARSr viruses where the amino acid sequence in the spike protein was very similar to SC2. These findings were remarkable for several reasons:
First, they showed that bats can harbor spike proteins that can directly infect human cells. Second, the nucleotide sequence was so similar that phylogenetic analysis could show that these bat viruses from Laos were the closest relatives of the SC2 spike. (Worth mentioning again that SC2 has a mosaic genome because of recombination. Different sections of the genome can have different evolutionary trajectories) Third, these Laotian bat viruses were even better adapted to bind human ACE2 receptors than the original Wuhan-1 strain of SC2.
On the other hand, no engineering team had the knowledge to create the RBD we observe in SARS-CoV-2. The 3D structure of SC2 spike protein was only solved months after the pandemic started, and structural biology is not something the WIV could’ve reasonably done in the first place (secret or not) since they lack this expertise. The best structural models any group of scientists had which could inform such engineering were based on the hACE2 receptor binding domain of the original SARS, which resembles the larger spike protein in shape, but uses a completely different binding mechanism involving other amino acid residues. It is possible to study binding affinity in existing sequences (and optimize them once the 3D structure has been solved) but it is currently almost impossible to design whole protein domains de novo for such high binding affinity.

To sum up all the lines of evidence, there is no room for doubt that SC2-spike and the affinity of its RBD to hACE2 receptors are of natural origin.
Was the furin cleavage site at the Spike S1/S2 position inserted?
There is no conceivable way how engineers could have designed the observed, but hitherto unknown, mechanistic synergy purposefully.
The furin-cleavage site (FCS) within the spike protein of SC2 has been a topic of much speculation, key among which is that it too is supposedly ‘unnatural’ and must’ve been introduced by researchers. An FCS is a short amino acid motif in the viral spike protein which comes in different shapes, but shares as a consensus a sequence of polybasic arginines (RR-X-R), which can get recognized by specialized proteases to then cleave the spike protein at this position. Spike-cleavage is a necessary step in viral processing and can happen inside or outside of the cell, and the FCS in SC2 has been implicated to increase virality and transmissibility by several studies.
The main arguments for the assertion that the FCS in SC2 is ‘unnatural’ are that no other currently known Sarbecovirus harbors an FCS at that position and that virologists have experimented with introducing FCS motifs, for example in SARS1 spike in 2006, (all in the open and without need for sensationalized ‘secret DARPA GoF proposals’) to study viruses for at least a decade.
I’ve already written about why this specific FCS in SARS-CoV-2 is highly unlikely to have been purposefully designed. To reiterate quickly:
The FCS in SC2 is highly uncharacteristic from an engineer’s perspective. Its sequence motif is inefficient (why introduce something that might not work when it is just as easy to insert a perfect motif?), it has an extra (not polybasic) proline spacer (which risks destroying protein function by itself as an alpha-helix breaker); and the whole motif opens up the surrounding domain structure for glycosylation (which might obstruct cleavage site recognition by the TMPRSS2 protease).
So all in all, these seemed like very odd, self-defeating, and avoidable design choices to me (and virologists come to the same conclusions).
Yet the so far best evidence against engineering comes from a functional study investigating the QTQTN motif directly ‘upstream’ of the FCS. The QTQTN motif is an uncommon sequence feature in CoV spike proteins, and determines how tightly the loop harboring the FCS is bound to the spike protein, thus regulating how well the TMPRSS2 protease has excess to it. Furthermore, the QTQTN motif is also glycosylated and loss of that glycosylation impairs viral replication. Researchers showed beautifully that there is an intricate functional interplay between the FCS, the loop length, and glycosylation: These separate but co-dependent elements work together synergistically and all elements are ultimately required for efficient viral replication and pathogenesis.
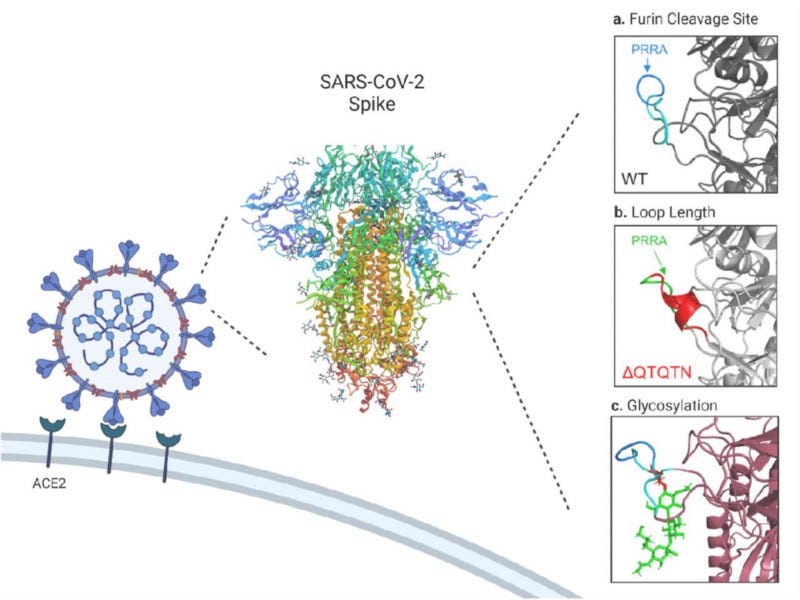
Disruption of any of these three elements attenuates SARS-CoV-2, highlighting the complexity of spike activation beyond the simple presence of a furin cleavage site. — Vu N.M. et al., bioRxiv, 2021
There is no conceivable way how engineers could have designed the observed, but hitherto unknown, mechanistic synergy purposefully. These synergistic interactions with genetic elements from the natural backbone were only discovered after the pandemic started. One cannot design what one does not know about. So the only remaining option for engineering scenarios requires that scientists might just have ‘lucked into this synergy’ by chance, using a viral backbone (we have no reason to believe they had) that featured a synergistic mechanism that made their (unnecessarily) crippled FCS increase fitness for human transmission despite the odds. What a perfect cocktail to stumble upon in the lab! I let you judge for yourself how probable you find this. (Worth mentioning also that SC2 loses the FCS in laboratory cell culture systems because of faster kinetics)
On the other hand, it is perfectly coherent with and expected from natural selection to gradually co-evolve the observed synergies. Anybody who ever studied molecular biology will tell you that these non-linear, cross-element synergies and co-dependencies are everywhere in evolved life, and for most, we have barely even scratched the surface to discover how they interact with each other.
Anyways, to bring this to a close: It would be shocking if the FCS was purposefully designed with the mechanistic synergy in mind, and awkward, because, despite our justified outrage, we then might also find ourselves hard-pressed to consider nominating this brilliant feat of engineering for the Nobel prize. (Okay, enough with the jokes)
Technically, engineers could have ‘lucked’ into this synergy which makes their oddly crippled FCS walk again, but I find this special pleading for miracles unrealistic. So we can be highly confident that the FCS was not engineered.
Why have we not yet found FCS motifs in other SARSr-CoVs in bats?
Here, there is some uncertainty left but multiple scientific explanations are on offer:
First, the simplest explanation could just be that we have not sampled enough viruses from the Sarbeco family. Before the Laos CoVs with their ‘human ACE2 optimized’ RBDs were discovered, we just had not yet observed such a feature. Again, the viral diversity in the wild is huge and we keep underestimating what genetic elements it could bring forth.
Second, while it is true that so far, no strong evidence for FCS in the Sarbecovirus family has yet emerged, these cleavage sites are not unique by any means; in fact, they independently develop all the time in the wider coronavirus family (and in other viruses like Influenza as well). The wider CoV family tree is sprinkled with FCS sites at the S1/S2 position.
This is not surprising, because FCS are very short sequences (4 amino acids) and these polybasic cleavage motifs will happen over and over just by chance through recombinations, insertions, or mutations. Most FCS motifs are likely acquired de novo and just don’t stick around if selection pressures do not favor them.
Furthermore, there is extensive evidence that the S1/S2 boundary region of the genome is a highly variable region in all CoVs and more susceptible to genetic alterations than the wider genome through point mutations, insertion, and recombination mechanisms.
The molecular mechanisms again are technical, but they have to do with RNA secondary structure & viral RNA polymerase slippage sites around the S1/S2 site, disrupting the replication machinery just enough to increase the odds of error or give opportunities for other ‘sequence’ pieces to be integrated at this position (potentially through template switching).
To support this notion of a ‘higher dynamic’ at the S1/S2 location, a recent pre-print of genomic studies of SARSr-CoVs in European bats found some indications that FCS at this location might pop in and out of existence in Sarbecoviruses at very low frequencies in a population. More studies will certainly shed some light on this issue, we just have to be patient.
Third, the same study of European bats also provided definitive evidence of proto-FCS sequences (polybasic amino acid motifs which are 1 nucleotide mutation away from becoming functional FCS). These findings are interesting because they allow for the possibility that a functional FCS in Sarbecovirus spike proteins might not only be evolutionarily ‘neutral’, but actually maladaptive (having them comes with a reduction in viral fitness & are therefore selected against).
Albeit maladaptation is pure speculation at this point, it is worth sketching out this argument from selection pressures as an example of how nuanced narrowing down the mechanism can be. The viral spike proteins in CoVs are under enormous selection pressures, because their shape defines not only what cells can be targeted, but also how efficiently the viral genome gets funneled into cells. Small changes in the spike protein sequence can come with a big fitness detriment, or expand the cellular tropism in unexpected ways.
The best real-life illustration we have of this is when looking at the different cell entry mechanisms between the spikes of SC2 (wild type) and the SC2 Omicron variant in humans. As part of its entry mechanism, the SC2 spike gets pre-cleaved at the S1/S2 FCS by the cell-surface bound TMPRSS2 protease, allowing the spike protein to open up and release the viral genome into the cell. Omicron spike (because of structural changes caused by its mutations) is inaccessible by TMPRSS2, cannot get cleaved outside the cell, and thus the whole viral particle gets swallowed into the cell through endocytosis before the viral genome gets released into the cytoplasm. These different entry mechanisms caused by mutations in the spike protein have wide-reaching consequences. Omicron reportedly displays a different cellular tropism (upper respiratory tract) than SC2 because expression levels of TMPRSS2 and general endocytotic capacities differ between human cell types.
Coming back to ‘missing’ FCS in bats:
Bat Sarbecoviruses might be more successful when not pre-cleaved outside the cell before infection. Consider again Section A, where we mentioned that CoVs in bats often go through the gastrointestinal system, not the lungs. Intestinal tracts are full of micro-organisms and chemical substances, making for a harsher environment than respiratory tracts. Pre-cleavage of spike outside the cell in the intestines might expose the viral genomes of Sarbecoviruses to damage, or interfere in other ways with cell fusion or entry, and thus could incur a fitness cost which ultimately causes selection against FCS-containing Sarbecoviruses.
Again, negative selection pressures for bat FCS are speculative at this point, but it might not be too far-fetched. MERS-like CoVs in bats are predominantly intestinal viruses and the closest bat relative of the MERS-spike, HKU4 spike, has no functional FCS, yet the MERS-CoV circulating in camels and jumping into humans has.
It is reasonable to expect that the moment environmental selection pressures change (e.g by the virus jumping hosts), a proto-FCS in Sarbecoviruses can quickly evolve to become functional cleavage sites in the new host through single point mutations.
Alternatively, de novo FCS acquisitions through recombinations or insertions, or low-abundance FCS-containing viral lineages would also grow out in these new host environments if selection favors them. That is the point of natural selection; once a genetic feature becomes adaptive, it will be maintained.
So while the ultimate answer to the ‘missing FCS’ in Sarbecoviruses is still not solved (or it might not even be real in the case of undersampling), we know that the flexible S1/S2 region in spike proteins is an error-prone spot of the genome favoring high genetic diversity, we know that multiple routes to acquire an FCS de novo exist in nature, and we know that FCS motifs evolved frequently and independently in the wider CoV family tree. Lastly, we know that once environmental selection pressures favor a genetic element (e.g through jumping hosts), viral lineages containing this feature will grow out to become dominant.
Conclusion Section B:
Every genetic feature of SC2 occurs over and over again in nature. SARS-CoV-2 has many closely related batCoV family members with partly identical sequences throughout every part of the genome. The vast viral diversity of CoVs in the wild allows for a variety of spike proteins with a high affinity to hACE2 to develop naturally.
The only plausible scenarios remaining for a lab leak would entail the collection (and subsequent escape) of a naturally evolved virus that was already ‘pandemic ready’ before it ever saw the inside of a lab.
A remaining area of uncertainty is why no other bat Sarbecoviruses have yet been found with an active furin-cleavage site. It is possible that we will find them with more sampling. Another possibility is that functional FCS in Sarbecoviruses are either neutral or selected against in bat’s cellular environment. There is no uncertainty that FCS motifs can be created de novo, either as a whole through recombination, insertions, or by single mutations activating an existing proto-FCS and those viral lineages would grow out once the host environment favors them. Multiple natural mechanisms are plausible and consistent with the evidence.
Overall, there is nothing extraordinary about the SARS-CoV-2 genome, just a ‘lucky’ combination of genetic puzzle pieces that made the virus sufficiently capable of establishing human-to-human transmissions once it got the chance.
It is also prudent to assume that there are many more of these ‘lucky’ combinations with dangerous genetic features already out there in batCoV reservoirs. A reservoir of ‘pandemic-ready’ viruses, so to speak, if we were to give them the opportunity.
By comparison, there is no evidence that a virus very similar to SARS-CoV-2 ever saw the inside of a lab. There are also no unnatural features in the genome, no genetic tags commonly used in research, no signs of gene editing or manipulation or culture. There is also no evidence of a close enough ancestor anywhere from which SARS-CoV-2 backbone could have reasonably been created through selection or serial passage. On the other hand, given the phylogeny of other Sarbecoviruses, there is no scientific uncertainty that the genomic backbone of any SC2 predecessor evolved naturally in the wild.
Therefore, no matter what potential lab leak scenario one considers, it has to start with the acquisition of a naturally-evolved unknown predecessor of SARS-CoV-2 by WIV scientists which was kept secret for years before the pandemic ever started. What makes this scenario even more unlikely is the arduous work going into isolating viruses from bat samples so one can grow them in cell culture. While the WIV had sampled a lot of bats and sequenced quite a few CoVs, they did not have vast arrays of different viruses in culture. In fact, in all the years of work, the WIV reportedly only ever managed to cultivate 6 CoVs, all related to SARS-1 which was of higher scientific interest.
Today, there is no evidence such a predecessor was ever found by WIV scientists, nor has it since been found by multiple large sampling endeavors from different research teams. Again, any cover-up of said ancestor would also likely have had to happen anticipatorily, many months/years before it allegedly ‘leaked’ and a potential need for a cover-up arose.
Lastly, critical genetic elements (falsely proclaimed ‘smoking guns’ for engineering), namely the RBD and FCS, have not only perfectly natural explanations for their occurrence, but they also harbor clear signs of being evolved in nature, rather than purposefully designed, contradicting further any engineering-based ‘heating up of a natural virus’ GoF scenarios. Taken together, there is not only any evidence for engineering, there is a dramatic amount of evidence against it.
The only plausible scenarios remaining for an accidental lab leak would entail the secret collection & cultivation (and subsequent escape) of a naturally evolved virus that was already ‘pandemic ready’ before it ever saw the inside of a lab. Or an asymptomatic infection of the field researchers working with bats, who then brought the ‘pandemic-ready’ virus back to the city without notice. If that scenario is more likely than the vastly larger number of locals, wildlife traders, or farmers bringing such a virus to the city of Wuhan is up for discussion.
But can any of these scenarios even account for the epidemiology we observed?
Section C) What are the likelihoods of a natural virus to start a pandemic at the wildlife market in Wuhan (a city that also houses a CoV research lab) compared to all other places in the world?
Before we can start speculating about geography, we first have to establish that Wuhan (and more precisely, the Huanan seafood market in November/December 2019) is the unequivocal place and time of where the first human-to-human transmission chains started. The heart of the pandemic, so to speak.
Phylogenetics and time estimations
Phylogenetics is a field of inquiry that studies the evolutionary history of genomes, building family trees based on genetic relatedness. These genetic family trees are useful when studying viruses; because they can both inform parent-offspring relationships as well as infer the time of emergence of the most recent common ancestor (MRCA) through mutation acquisition rates.
One peculiar detail about the early viral genomes that were collected between December 2019 and June 2020 is the fact that they all belong to either one of two distinct sub-lineages of SC2, termed ‘A’ and ‘B’, causing separate transmission chains in China and outside. Viral genomes of the ‘A’ lineage were found and sequenced later than the B lineages, but are thought to be ancestral because they differ by two nucleotide mutations (C8782T and T28144C) that are shared by related bat viruses.
The ‘B’ lineage accounts for the bulk of cases in the pandemic and all future variants of concern (Alpha, beta, gamma, delta, omicron) evolved from offsprings of the ‘B’ lineage. ‘B’ lineage genomes were also the first found in sick patients in Wuhan and have a firm epidemiological link to the Huanan seafood market (more on that later).
The fact that there were two distinct lineages early on is of critical importance, as this could imply a hidden community transmission for months (lineage A could’ve evolved into lineage B) before the outbreak at the Huanan market. Alternatively, the two lineages could have evolved in an animal reservoir from a shared MRCA before separately jumping into humans at the market. Option one is compatible with a lab leak scenario, option two is not.
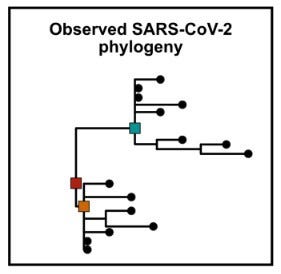
One way to probe these possibilities is at looking for evidence of ‘transitional genomes’ between lineage ‘A’ and ‘B’ in humans. Because mutational acquisitions happen step-wise, if one of the lineages is indeed the only direct grandfather of the other, a transitional genome would be the equivalent of the parent. If that parent were to be found in humans, it could point to asymptomatic spread before lineage separation. Initially, a small number of these ‘transitional genomes’ were reported, but upon closer inspection, every single one of them turned out to be a “false positive”. Contaminations (accidental mixes of A & B samples) or artifacts from sequencing or bioinformatic processing are quite common and not always easy to spot. No evidence of transitional genomes makes a single introduction less likely.
Additionally, a recent phylogenetic study could show that the hundreds of sampled genomes (either belonging to descendants of lineage A and B) create lineage trees quite distinct from each other (while being of low overall diversity) that their occurrence can hardly be explained by coming from a single introduction into humans. Both lineages A and B exhibit large polytomies (multiple sampled genomes descending from a single node on the phylogenetic tree, see below), which rarely occur when simulating a single introduction event as the start.
To put it in other words: There are 108 and 231 lineages descendent from the base of lineages A and B, respectively. Every viral genome acquires random mutations in different places, gradually propagating forward (and forming a clade) or dying out (when the transmission chain ends).
The assumption that hundreds of genomes acquired the same (lineage-defining C8782T or T28144C) mutations first and only then starting a separate polytomy with random mutations (which is the data we observe) is very unlikely (between 0,3–3,6% probability in simulations, see below).
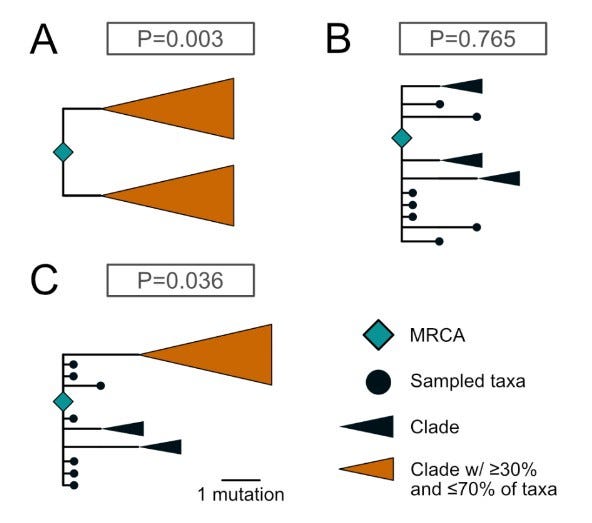
The epidemic simulations which best recapitulate the observed data suggest having multiple (at least two!) separate introductions into humans, serving as the starting point from which the two observed polytomies spread out.
One more important point on the likelihood of ‘multiple spillovers’ from an animal reservoir. People‘s intuition might lead them to believe that because one spillover is unlikely, two spillovers are way more unlikely. This is not always true when dealing with conditional probabilities. We have to consider that all the difficulties and unlikelihoods for zoonosis lie ‘upstream’ of the market. It is very rare to find an intermediate animal infected with a ‘pandemic-ready’ bat virus and bring it into close proximity with humans for weeks. Once these unlikely pre-requisites are fulfilled, however, multiple spillovers are extremely likely, because the infected animals are there spewing viral particles into their environment. To use an analogy from Joel Wertheim: “Humans failed to climb Mount Everest for hundreds of thousands of years. And then, in just one day, two people did”. Once humans reached a state of tech that made it possible to climb such heights, more humans are expected to do so. This is conditional probability. Or take a real-life example: Catching Covid-19 in late January as a US citizen was extremely unlikely (less than 1 in 10 million), SC2 hasn’t spread far yet, the virus wasn’t as infectious as Delta or Omicron variant and overall global case numbers were minuscule. Finding (at least two) random US citizens with no personal connection to each other being simultaneously infected would be almost impossible odds (if we ignored conditional probability). Now imagine looking on the Diamond Princess cruise ship in early January after a sick patient from Hong Kong entered it. (caused the odds from 1 in 10 million to skyrocket to about 1 in 4 passengers being infected) Finding multiple infected US citizens (who have no personal connection to another) on that boat housing hundreds of passengers becomes a virtual certainty. That is conditional probability.
Back at the Huanan market, it is likely that several zoonotic infections without sustained transmission were missed because they went extinct. We know from other pandemic simulation experiments that even in dense cities like Wuhan, a pathogen as infectious as SC2 will go extinct 70% of the time before infecting enough humans to establish stable transmission chains. This means that although we only have evidence for two separate zoonotic jumps, it is very likely that we missed several others from the same animal reservoir because asymptomatic transmission chains terminated early. Pekar et al. ‘s epidemic simulations offer a confidence interval from at least 2 up to 15 zoonotic jumps being consistent with the SC2 case numbers. Yet we only ever got to observe the lineage successes, not the misses (infections that went extinct). Similar to the Diamond Princess example, we have to assume that once the critical parameters for zoonosis were in place at the market, it would be odd to assume only two jumps happened.
One last example to drive this point home: We have already observed “multiple spillbacks of SC2 into humans” from infected minks and white-tailed deers, who became recent animal reservoirs for the virus. Multiple zoonotic jumps are common and expected from animal reservoirs once the pandemic-capable virus is present.
Another big part of the Pekar et al. pre-print used their epidemic simulations, phylodynamic tMRCA estimates with different roots, multiple index cases, and hospitalization dates to narrow down the most likely time window of when a zoonotic spillover happened using a previously developed model. I’ll not go into the technical details here because it would break the length even more. Read the paper or take the author’s summary:
Therefore, across multiple phylodynamics models, index case or earliest hospitalization dates, and epidemic doubling times, our results indicate that lineage B was introduced into humans no earlier than November 2019, and lineage A cross-species transmission likely occurred within days to weeks of the first event. -Pekar et al., bioRxiv, 2022
Alright, so to sum up:
Phylogenetic evidence and epidemic simulations indicate that two separate introductions of SC2 into humans happened close to each other somewhen in November/December 2019.
But where exactly did this happen?
Case epidemiology of hospitalized patients
One big problem with the disease epidemiology of SC2 is the large proportion of people who are either asymptomatic or not sick enough to be recorded. Only the increase in hospitalizations of severe pneumonia cases raised the alarm bells in Wuhan weeks after the pandemic was already spreading in the community. Finding and confirming SC2 infections weeks after the fact is necessarily incomplete by the nature of the disease and mostly requires detective work. This detective work by the Chinese CDC and WHO lead to the association of the outbreak with the Huanan market:
[…] retrospective clinical case-finding concluded that 55 hospitalized COVID-19 cases with symptom onset in December 2019 had a link to the Huanan market, out of the 168 for whom exposure history to this market was available (33%).
Since then, many lab leak skeptics and internet sleuths have leaped to these results to raise doubt about detective work that by its nature is operating with a lot of uncertainty. Ascertainment bias, missing cases, hidden hospitalizations, and purposeful cover-ups were insinuated with little evidence but great fanfare.
So how to ever get past unproductive conspiracism when uncertainty is so high? Well, with more exhaustive sleuthing done by scientists, and some impressive statistics, of course.
In a recent preprint, scientists used the maps in the WHO mission’s report on the origin of SARS-CoV-2 to extract latitude and longitude for most of the known COVID-19 cases from Wuhan with symptom onset in December 2019 so they could create density maps of all known cases, whether they had a link to the Huanan market or not (see below).
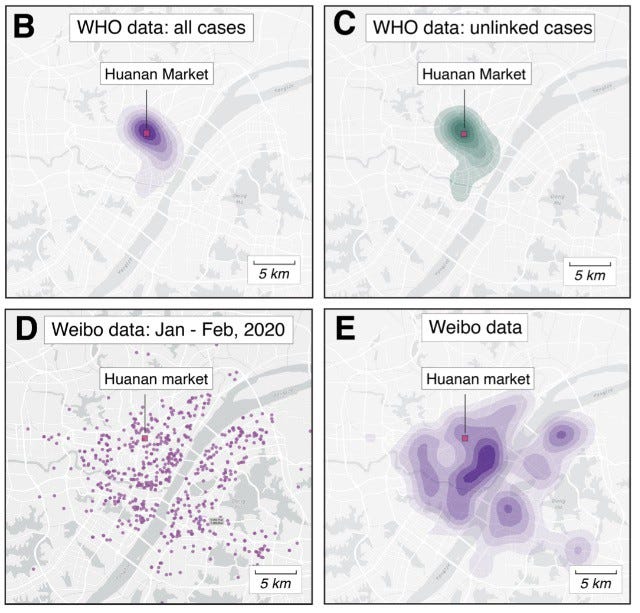
Importantly, even pneumonia cases that had no association whatsoever with the market (no work, travel, visits, or contacts there) still centered around the Huanan market and could not have been subject to ascertainment bias. The market was the only place in Wuhan where early cases had a clear association. There are no other epidemiological links to any other place in the city, other clusters only started forming later in Jan-Feb (Weibo data, shown above) and became more representative of the city’s population density.
It is worth mentioning here that the Wuhan Institute of Virology (the alleged ‘escape’ laboratory working on CoVs, not shown in map) is located on the other side of the Yangtze River, South West of the Dong Hu lake, more than 16km away from the Huanan market, and no sickness clusters of cases were shown anywhere close in December 2019.
Next to the clustering analysis and all the patients with a clear connection to the market, there are other pieces of evidence that put the Huanan market as a likely origin of the pandemic: Environmental swaps testing positive for SC2 and SARSr-CoV susceptible animals.
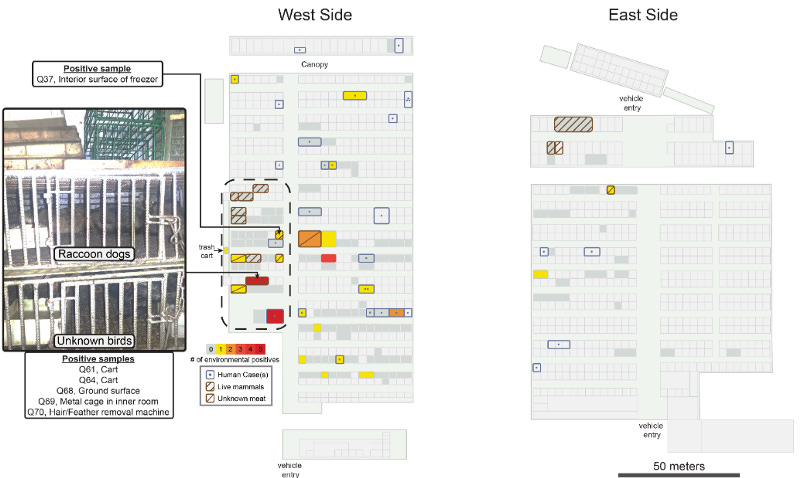
When the Chinese CDC came to the Huanan market, they took many environmental samples from inside the market; including surfaces in shops and from cages, as well as from equipment and sewage. From 828 samples inside the market, 64 samples (7.7%) were positive, especially the west side of the market showed higher contamination. The market is separated into two zones, eastern and western zone, with seafood and animals mainly sold in the western zone and livestock meat in the eastern zone (shown further below).
Now at the time when the Chinese CDC was investigating the Huanan market, they reportedly had not found any evidence of SC2 positive animals. Furthermore, they reportedly have not taken samples from many animal species scientists since found out had been sold at the market prior to the pandemic and up until December. There are many possible explanations for this. The animals could have been culled, sold, or removed by traders before the CDC came to inspect on January 1st. After all, many saw the resemblance to the SARS-CoV-1 outbreak in 2003 and might have been panicked to get rid of potentially infected animals. Another explanation might include Chinese scientists not being allowed to share information because of political pressure. The reasons are speculative, what matters is that there is (even photographic, see below) evidence for these animals being at the market at least until the 3rd of December 2019. Longnitudinal studies of animals at the market since 2017 and recovered inventory lists also make it quite clear that SARSr-CoV susceptible animals had been held and were sold at the Huanan market.

Among its life animals, the market held raccoon dogs, Asian and hog badgers, marmots, foxes, and bamboo rats, all of which have been shown to be susceptible to SC2, and in the case of raccoon dogs, were previously implicated in the emergence of SARS-1 in 2003. In a remarkable feat of detective work and statistics, scientists could show that the market areas with the highest viral contamination were overlapping with the stalls that sold life animals, including one shop that housed cages where the raccoon dogs were kept. While the animals were reportedly gone by the time the CDC arrived to take environmental samples, multiple environmental swaps in that shop came back positive for SC2, including the surface of the cages, the sewage under the cages, the fridge, and a defeathering machine (environmental swaps Q37, Q68–70). No human infections have been reported in or near this stall, further indicating that the environmental contamination had an animal origin.
The epidemiology and location data clearly point towards animals at the market. We have already explained that the phylogeny points towards multiple zoonotic jumps and not a single introduction followed by a super spreading event (as some researchers had previously suggested).
Another real eye-opener against superspreading came from a recent pre-print (Gao G. et al., researchsquare, 2022) from the Chinese CDC, when the full-genome sequencing of environmental samples taken on 1st of January 2020 identified an SC2 genome belonging to lineage A. The environmental swap (sample #A20) came from a glove on the western side of the market.
Lineage A had no business being there at the market if the market association was just due to a lineage B superspreader event. While infected humans can walk around, obscuring where exactly they got infected, environmental swaps are pretty location-bound. If lineage A could be found at the market, it’s because one of the animals or wildlife traders was shedding large amounts of lineage A virus into their environment. Furthermore, a super spreading event is also inconsistent with the geospatial findings that both lineage A and B still center around the Huanan market, and of course with the phylogenetic analysis indicating multiple spillovers.
The finding of lineage A from environmental swaps at the Huanan market both supports the independent epidemiology and phylogeny analyses and ties the knot on establishing the Huanan market as the place of origin of the SC2 outbreak in humans.
Why the city of Wuhan?
The fact that Wuhan, a city that houses a BsL-4 CoV research lab, became the epicenter of the SARS-CoV-2 pandemic has been the driving force behind the lab leak hypothesis. Many public figures and influencers love to proclaim the apparent obviousness that from all possible cities in China, the odds that an outbreak would fall on Wuhan are so minuscule as to not be a coincidence. Given that all the evidence points to the Huanan market, I am not sure how relevant this coincidence ultimately is, but I think it certainly was a legitimate question to prompt further inquiry.
So I’d like to just raise a few considerations to better estimate how big a coincidence it would really have to be. Epidemic simulations showed that a pathogen as infectious as SC2 would likely not cause an epidemic if it infects humans outside of dense population centers. Humans would either not meet enough people to infect, driving transmission chains to extinction, or would not travel enough, building ‘village’ immunity after everybody susceptible got infected and acquired immunity.
We can expect SC2 epidemics would only happen in population-dense cities. There are still 102 cities in China hosting a million inhabitants or more, so the ‘coincidence’ of Wuhan might be in that ballpark without further information. Since Wuhan is the 10ths biggest and 7ths densest city in the country, maybe the zoonotic opportunities are a bit higher than any generic city in China, but not too much.
However, Investigations into wildlife farms in Hubei province (the province housing Wuhan) showed that wildlife breeding was widespread in Hubei, some farms are even near bat caves, and that supply chains from these farms run into big cities (including Wuhan and Foshan). These practices have been encouraged by the Hubei government specifically and might not be as common in other provinces. I have already mentioned a study that showed that in the leadup to the SC2 pandemic, pork supply in China was decimated causing prices to skyrocket. Hubei was among the provinces hit the hardest with increasing pork prices, which led to alternative sourcing of meat, including from wildlife and fur farms.
How much these regulatory and economic differences between wildlife trade and factory farming in various Chinese provinces matter is unclear; the fact that (likely ancestral lineages of) SARS-1 were also found in animals on Hubei farms near Wuhan (far away from the bat reservoir in Yunnan and the original outbreak site in Foshan) certainly allows for speculations that there are local factors in Hubei that facilitated SC2 emergence in Wuhan.
So where does this leave us with all the people claiming that a city with a CoV lab having a CoV outbreak would be a peculiar coincidence? They are not entirely wrong, at least prima facie, there is currently no strong evidence to suggest that similarly population-dense cities with wild animal markets like Foshan (SARS-1) or Chengdu (closer to Yunnan bats) are not equally as likely to be centers of a future CoV pandemic. More investigations could help (see next chapter).
Without further evidence, the odds of a novel CoV pandemic arising in Wuhan out of all other places in China might be somewhere in the 1:10–1:100 ballpark. That is low, but certainly not unimaginable.
But how much does this geographic coincidence really matter given that the same logic could’ve been applied to the SARS-1 outbreak in Foshan in 2003? Geographic coincidences are not a critical factor in singular events. It could just be survivorship bias. While it is intuitive, it might be an ultimately flawed assumption.
The relevant ‘Bayesian likelihood analysis’ is not about the city. It’s about how likely it is that from all possible places where a virus might break out within a city, all the cases, environmental samples, and epidemiological lines of evidence point toward a wildlife market housing animals known to carry CoVs, but nowhere else? Nowhere near the WIV. Nowhere near nursing homes, restaurants, churches, theaters, or other community centers that had been superspreader events in other countries. Ah, and of course the ‘coincidence’ that the market was not only the center of the emergence of one, but at least two separate lab leaks about a week apart. I’ll let you do the inference.
The ‘coincidence’ that two separate lab leaks show up first at a wildlife market, the only place in the city that we know housed precisely those wild/farmed animals which serve as reservoirs for SARSr-CoVs, certainly strains credulity without supporting evidence.
Conclusion Section C:
The geographic coincidence of a coronavirus outbreak happening in a city that houses a research facility specializing in exactly those viruses is small. To date, no dispositive evidence has surfaced that would make Wuhan a much more likely place of zoonotic spillover than other major cities with similar wildlife markets, like Chengdu or Foshan. Models predict that a virus-like SARS-CoV-2 has high odds to run itself out, rather than cause a pandemic, even in very dense cities, so we ought to be careful not to over-emphasize the geography of a singular event, which might come down to opportunity, luck and survivorship bias. After all, that SARS-1 broke out Fushan had likely little to do with Fushan but bad luck; scientists had since discovered animal reservoirs for SARS-1 also around other cities, including in Hubei province animal farms and right outside Wuhan.
Every single piece of epidemiological evidence points to the Huanan market as the place where SC2 emerged.
By comparison, the phylogenetic finding that lineage ‘A’ and ‘B’ are representing at least two separate introductions, rather than one lineage transforming into the other in humans, is difficult to explain with any lab leak scenario. The finding of lineage A environmental samples at the market, the clustering of lineage A cases around the market, and epidemic simulations also discard a super spreading event as a plausible explanation for the market association.
The likelihood of (at least!) two separate pathogen escapes of two almost identical viruses from a lab is vanishingly small. And before the smartypants of you will say: But what about conditional probability here? Shouldn’t it apply to the lab leaks too that if one happened, the second becomes more likely too? In principle, yes this can be true. The difference is that for zoonosis, all the difficult and unlikely steps are ‘upstream’/before the ‘pandemic ready’ virus finds itself at the market, from there on, it’s expected and likely to spill over multiple times. Whereas for lab leaks, the difficulties and unlikely events begin ‘downstream’ after the virus is in the lab. (This is already assuming the WIV had the virus, which in itself is not supported by any evidence and is unlikely given how few CoVs they ever managed to isolate, but okay, let’s play devil’s advocate)
First, consider the limited opportunity for these leaks to happen, the limited amount of people with access to these viruses, and the lack of scientific rationale to even have two almost identical viruses in a culture that only differ in two random nucleotide mutations in uninteresting regions of the viral genome. Two leaks happening almost simultaneously (a week apart) is less likely, not more.
Second, the geographic emergence of both lineages at the Huanan market (but nowhere else) can hardly be explained with (at least!) two distinct lab leaks at the WIV ten miles away, as we would expect pneumonia case clusters all around the lab for both lineages. And if not around the lab, why not around the countless nursing homes, churches, theaters, or other community centers Wuhan has to offer which are prone to super-spreading events?
The ‘coincidence’ that two separate lab leaks show up first at a wildlife market, the only place in the city that we know housed precisely those wild/farmed animals which serve as reservoirs for SARSr-CoVs, certainly strains credulity without supporting evidence.
Can any lab leak hypothesis really account for the observed data?
Chapter 3: What next?
Remaining uncertainties
In this article, I highlighted how multiple lines of scientific inquiry strongly point towards a natural origin of SARS-CoV-2 via emergence at the Huanan market in Wuhan and that such a zoonotic origin hypothesis is internally consistent with any and all pieces of scientific evidence. Please consider that no single article can summarize all valuable scientific contributions that scientists have made in support of investigating the origin question, and for zoonosis specifically. It is a large and growing body of evidence. My contribution to this topic was to focus on explaining key scientific arguments for non-experts and also accurately present the conclusions scientists reached in their work by considering that body of evidence. While I did go to great lengths to avoid misrepresentations, I cannot always control how my words will be interpreted. If there are uncertainties arising from my simplifications, omissions of brevity, or bad analogies, I advise you to first consult the primary literature for clarification rather than presume there is an obvious mistake in the science or reasoning of scientists. At this point, the scientific consensus on key issues surrounding the origins of SC2 is pretty clear. However, this does not mean that there are no more questions to be asked, or no more mysteries to be solved.
For example, whatever happened before the outbreak at the Huanan market has yet to be worked up by scientists. Figuring out which animals, whether they are associated with farms or wildlife, and how they connect to bats reservoirs is still unclear. There is serological testing of wildlife traders and farmers to be done, and future risks to be assessed. This work could also better elucidate whether Wuhan was more prone to have had an outbreak like SC2, or whether it was a coincidence that it happened there and not in other cities with wildlife markets, like Fushan for SARS-1. Unfortunately, in no small part because of the lab leak controversy and geopolitics, Chinese cooperation on these investigations is currently slow, to the frustration of many scientists.
Many place their hopes in the WHO’s SAGO mission, but skepticism remains whether this climate of assigning blame can really be left behind. Also, conspiracy fantasies will not suddenly vanish just because they are not based on scientific evidence, as the anti-vax movement has shown.
This brings me to my next point:
What to do about lab leak-related conspiracism?
What can be asserted without evidence can also be discarded without evidence.
There is a vast literature of extant evidence that could be used in this already too-long long article to further debunk many baseless arguments and conspiracy theories surrounding the lab leak hypothesis. But do scientists really need to engage and debunk every unserious claim advanced by non-expert influencers with science?
What about claims that SC2 cases circulated in New York, Italy, or France months before the pandemic started in Wuhan? After all, weren’t there some serological survey from pre-pandemic blood samples taken which found some “evidence” SC2 infections? (False positives usually caused by the known cross-reactivity of other hCoVs with the ELIZA assay or PCR contaminations had been known for a long time now)
Or what about the never-funded, but surely ‘nefarious’ DARPA grant proposal? (Couldn’t have produced SC2 (as we now hopefully understand) even if the work was ever performed)
And hey, what about the military games that happened in October in Wuhan? (What about the moon constellation in October or anything else that is irrelevant?)
There will always be people with wild theories and no scientific evidence to back it up. In the age of chaotic online discourse, we would be wise to remember that what can be asserted without evidence can also be discarded without evidence. It would be good to remember that, but this would not solve all problems. We are also past the point where claims of ‘alternative scientific explanations’ for individual datasets or insinuating problems with ‘missing data’ can move the needle on zoonosis. Multiple lines of evidence would have to be proven wrong to change the consensus emerging from the body of evidence. Cherry-picking data out of context is scientific malpractice but hard to spot for non-experts. Scientists have currently no recourse against malicious misrepresenters of science but to call out BS in public whenever they see it, but this exposes them to being dragged into a fight for audience attention they cannot win. We, the public, have to be more confident in the scientific consensus, and not get distracted by fanciful alternative explanations that seem sound to our intuition.
In the end, we would want scientists to have better things to do for us than debunking conspiracies or sensationalist speculations that have no supporting evidence (or cannot be disproven with evidence in the first place). Science is about finding and navigating competing hypotheses that best explain reality. It is about being exhaustive, considering all the available evidence, and pointing out where competing theories fall short to do so. The current crowdsourcing of misinformation on social media is something we all struggle with at the moment, not a problem of insufficient science communication. Scientists have stepped up dramatically to do more science outreach in the pandemic, but they cannot do it alone.
It is up to society to support scientists so they can focus on science, not put the burden on them to solve online conspiracism around their topic of expertise. We are exposing them to public hatred for refuting sensationalist stories and then leaving them to fend for themselves and their reputation against political actors. These are terrible incentives.
I hope this article made clear that the body of all available evidence is pretty unequivocal in showing that zoonotic jumps of CoVs are common and pose a high risk to humans, that the SC2 genome is not engineered but evolved naturally, and that the pandemic started at the Huanan wildlife market, most likely through at least two distinct lineage introductions.
And while the door is always open to new evidence to be discovered, any lab leak theory worth paying attention to must be consistent with the whole body of evidence, not offer selective ‘what if’ fantasies that might explain one part of the evidence, but be contradicted by another. None of the lab leak scenarios currently advocated for are fulfilling these criteria, even if we allowed for all insinuated (but unsupported by evidence) cover-ups to have taken place.
Therefore, until a scientifically plausible lab leak hypothesis scenario can be formulated, I doubt that any productive engagements can be had with the fanatic core of lab leak advocates. Take these bitter grapes also as the hard-learned lesson from someone who has been sympathetic to the possibility of a lab leak and engaged in many good faith discussions with sensible lab leakers for over a year now.
A better way forward
In life, we often don’t have perfect information and yet still we have to make decisions and solve problems. We don’t know for certain that we will not have a deadly car crash the next time we drive, yet from a combination of knowledge about our driving skills, weather conditions, and overall car fatality rates, we can reasonably assess that we will most likely not die the next time we get in the car, so we do. This state of ‘actionable certainty’ is what is required to function in life, and navigating any collective action in society is maybe not that different. We all agree that risking another pandemic by doing business as usual is probably not the greatest idea, yet the controversy around the origins of this pandemic has distracted us from taking real action to prevent the next one, even made it harder to investigate pandemic risks and act collectively against them.
The real enemies to thwart are the devious biological algorithms that feast upon our bodies, not our fellow humans.
Scientists have provided ‘actionable’ certainty that this pandemic, like many before, has had its roots in nature’s relentless GoF laboratory, and most likely ‘leaked’ into humans because of our unwise practices with animals and other unforced errors. From encroaching into wild habitats to factory farming, from animal cruelty for profit in population-dense places with high mobility to insufficient pathogen monitoring and slow political response times, a lot of things have to go wrong together to allow for a pandemic to happen. This is where the bulk of our attention and resources should go to prevent the next pandemic.
Pandemic preparedness does not preclude or absolve any discussion on biosafety, research transparency, or oversight. Quite the contrary. Although we can be reasonably certain that this pandemic resulted from a zoonotic spillover of a natural virus from an animal reservoir, we should be concerned about the possibility that the next dangerous pathogen might escape from a lab. Biocontainment concerns are taken seriously and are shared by are all scientists I talked to, and it is not surprising why. Scientists who work with these pathogens worry about them all the time. Since 2020, we all have experienced what living entails while dealing with the presence of a dangerous pathogen. I doubt that false ‘artificial’ origins of SC2 need to be conjured up to make the case for biosafety, nor for scientists to take research hazards seriously. The pandemic has been a teaching moment to show all of us, including scientists, where we ought to do better to reduce risk. For example, enforcing more transparency and thorough oversight in ePPP research is one thing to discuss, better-equipped facilities and more training of scientists is another. There is no reason why BsL-4 labs have to be in dense cities, so moving them to remoter places might be a good suggestion too. However, this is the easy part of a long list of things to do.
Surveillance of animal reservoirs, education for people interacting with animals, enforcement of wildlife trade/factory farming regulations, monitoring of zoonotic hot spots, and establishing fast-acting pandemic playbooks are the necessary but more difficult parts that are currently neglected by society. Long-term preventative measures, like vaccinating susceptible animals or reducing economic incentives for countless risky human-animal interactions globally are also in dire need to be discussed (& even more difficult to implement without reducing global and local economic inequalities first). Still, working towards these measures is what’s required to reduce the risk of a repeat of the SC2 pandemic.
What truly worries me is not the enormity of the task, but the inertia of society to get started. It’s been more than two years. I fear that all lessons this pandemic taught us are wasted when supposedly only scientists have to heed any call to action, as most lab leak grifters dominating online discourse will have society believe. Stopping ‘risky’ research to prevent pandemics like SC2 is a naive fantasy. Simple solutions to complex problems often are.
Societal problems need societal actions, not false promises. Easy fixes like ‘shutting down GoF virology” are knee-jerk reactions that will likely do nothing to move the needle on actual risk while potentially harming preparedness for the next pandemic. Or maybe they do move the needle a tiny bit. Of course, we can and ought to discuss research practices, maybe the benefits of ePPP virology are too small to be worth the risks, but there is no discussion that lab safety is at best a minor sideshow to a much larger challenge of pandemic prevention and preparedness. Nature is a gigantic GoF laboratory with countless dangerous experiments running every day. Natural viruses are also leaky as hell and we humans are aiding & abetting pandemic pathogens to reach us through our actions and unwise practices. Can we start transcending false narrative simplicity in favor of reality?
We ought to come together on this one. We have to remember that nobody, no ‘lab leak’ enthusiast nor ‘zoonotic’ origin proponent, is on ‘Team Virus’ or rooting for the next pandemic. The real enemies to thwart are the devious biological algorithms that feast upon our bodies, not our fellow humans.
All I want is for humanity to start acting that way too.
Epilogue
There is a lot going on in the world right now and I cannot pretend the Russian invasion of Ukraine has not shifted the world’s focus and priorities, and rightfully so. Decades of misinformation have contributed to gradually shaping a nation of 150 million to become cynical or ignorant enough to allow a kleptocratic madman in power to usher in a new dark age of war and nuclear escalation.
Yet the seeds of cynicism and ignorance fester in all societies, autocratic or not. They creep in wherever we allow the inherent authority of science to get pushed aside by motivated actors pursuing their own agendas. Science has an important function in society, it establishes a shared reality through shared facts. The online age has made it too easy to perceive reality in whatever way feels intuitive to us, to pick facts that suit our worldview and ignore the rest. To live in echo chambers. Yet the perceived reality we choose to live in is a fickle and malleable thing, especially when merchants of doubt and cynicism have taken over public discourse, pulling us back into the darkness of our own irrational fears or tribal desires. No good decisions can come from fantasies. No clarity can be won from make-believe. Ignoring reality whenever it is personally convenient or profitable is a recipe for disaster. Allowing liars to run rampant with misinformation for a long time is not only a problem for the Russian population.
“It is far better to grasp the Universe as it really is than to persist in delusion, however satisfying and reassuring.”
― Carl Sagan, The Demon-Haunted World: Science as a Candle in the Dark
Scientists only know how to fight fiction with facts. They cannot win shouting matches with professional grifters who capture our attention for a living. It is up to us, everyday humans like you and me, to lend a hand.
The only defense we currently have against crowdsourced misinformation and propaganda is to uphold the role of science in society. It lies in our own willingness to award our trust, or at least our attention, to the fruits of the scientific process.
References:
The very fact that you are closed minded and refer to the possibility of a lab leak as "conspiracy" proves that you are not a scientist.
"Scientists take biosafety and public concerns seriously"
Stop lying.
https://www.propublica.org/article/near-misses-at-unc-chapel-hills-high-security-lab-illustrate-risk-of-accidents-with-coronaviruses
https://www.dailymail.co.uk/health/article-11323677/Outrage-Boston-University-CREATES-Covid-strain-80-kill-rate.html